The worldwide increase and proliferation of drug resistant microbes, coupled with the lag in new drug development, represents a major threat to human health. In order to reduce the time and cost for exploring the chemical search space, drug discovery increasingly relies on computational biology approaches. One key step in these approaches is the need for the rapid and accurate prediction of the binding affinity for potential leads.
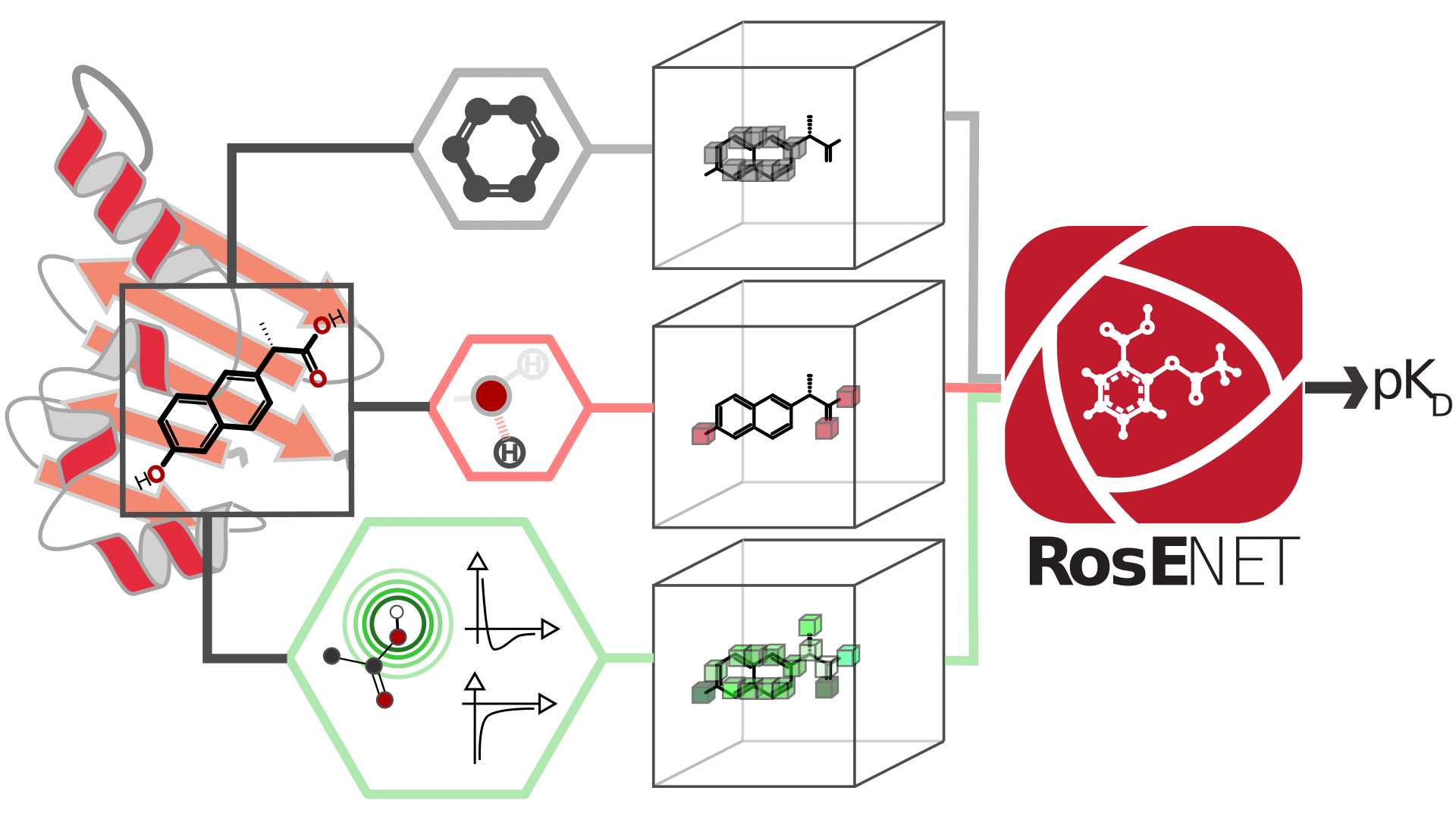
Here, we present RosENet (Rosetta Energy Neural Networks), an ensemble of three-dimensional (3D) Convolutional Neural Networks (CNNs), which combines voxelized molecular mechanics energies and molecular descriptors for predicting the absolute binding affinity of protein–ligand complexes. By leveraging the physicochemical properties captured by the molecular force field, our ensemble model achieved a Root Mean Square Error (RMSE) of 1.24 on the PDBBind v2016 core set.
We also explored some limitations and the robustness of the PDBBind data set and our approach on nearly 500 structures, including structures determined by Nuclear Magnetic Resonance and virtual screening experiments. Our study demonstrated that molecular mechanics energies can be voxelized and used to help improve the predictive power of the CNNs. In the future, our framework can be extended to features extracted from other biophysical and biochemical models, such as molecular dynamics simulations.