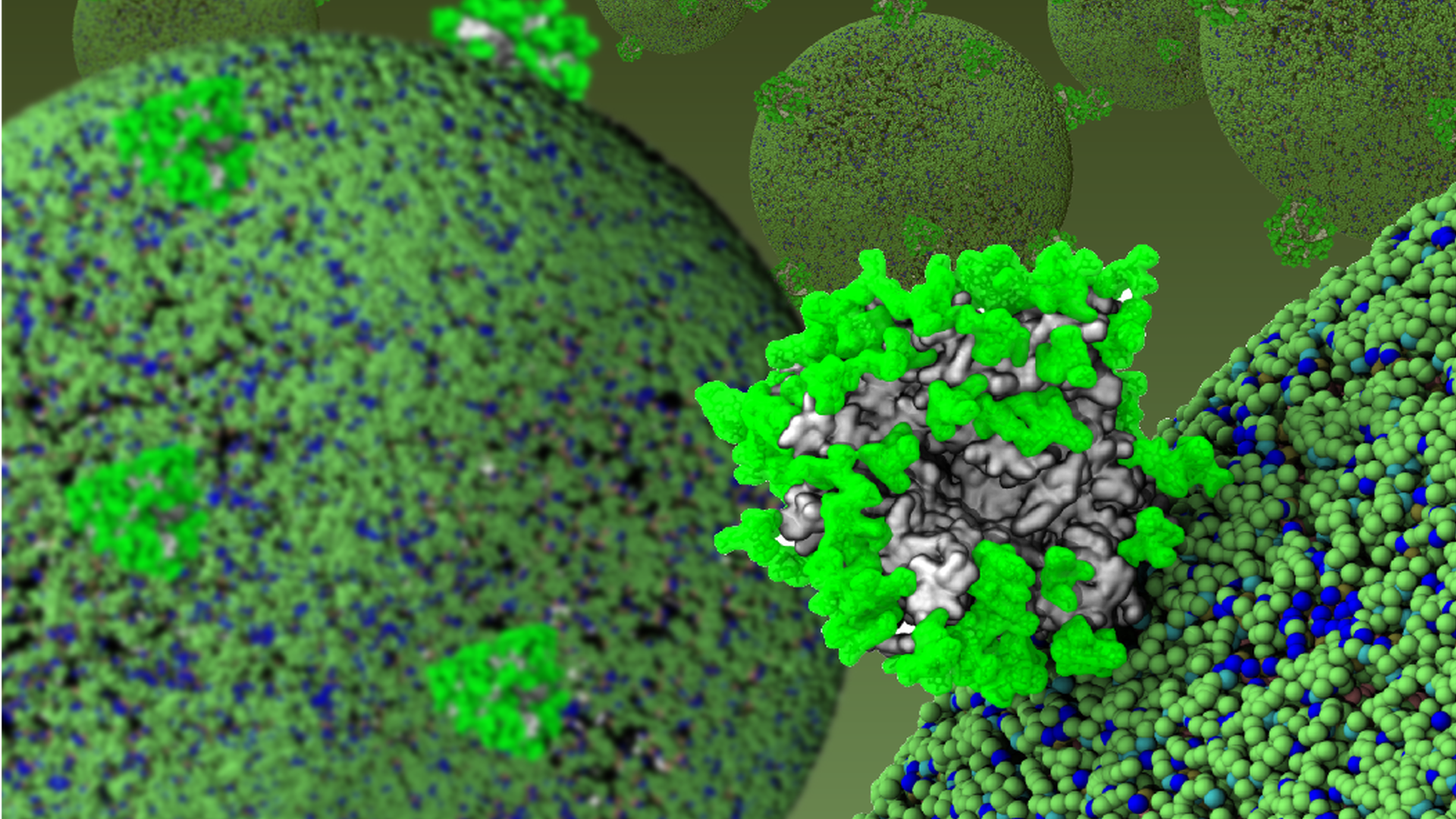
The HIV-1 envelope (Env) is one of the most glycosylated proteins known, with roughly half its mass comprising host-derived N-linked glycans. Little is known about the structural dynamics of these N-linked glycans and how they coordinate to shield the Env-protein surface from potentially neutralizing antibodies.
At the Vaccine Research Center (VRC) at the National Institutes of Health (NIH, USA), I pioneered the use of molecular dynamics simulations and graph theory to investigate the dynamics of the HIV-1 Env glycan shield. The fully glycosylated HIV-1 Env is a highly complex system and required the development of several programs to set-up the molecular dynamics simulation. My work not only allowed the first visualization of the HIV-1 glycan shield (something that had eluded the HIV-1 community for over a decade), but it also provided us with structural proof that all broadly neutralizing antibodies must account for the presence of the glycan shield. A network analysis of the interactions between glycans showed a collective behaviour and that they are organized in micro-domains, which could have important implications for CD4 recognition and broadly neutralizing antibody recognition.
In addition, I employed molecular dynamics simulations to characterize and model the binding of antibodies to the HIV Env trimer. My analysis of the accessibility of the fusion peptide and distance from the viral membrane demonstrated that the fusion peptide could be a site of vulnerability to neutralizing antibodies. I modeled the binding mode of a CAP256-VRC26.9 broadly neutralizing antibody. The model was fully consistent with available cryo-EM and HDX-MS data. It is currently being used for structure-based design aimed to improve its neutralization breadth. This work was reported in the journal Cell and the glycan shield visualization appeared on its cover.